- Nature
- 497,
- S19–S20
- doi:10.1038/497S19a
- Published online
Sleep disturbances may be an early sign of neurodegenerative diseases — but could sleep deficits cause these conditions in the first place?
Jae-Eun Kang was using a new microdialysis technique to measure how levels of soluble amyloid-β protein in the mouse brain change in response to physiological stress when she noticed something odd. It's thought that the level of soluble amyloid-β correlates with the eventual formation of amyloid-β plaques in the brain, one of the hallmarks of Alzheimer's disease. But Kang found that the protein's concentration seemed to peak during waking hours and fall as the mice slept.

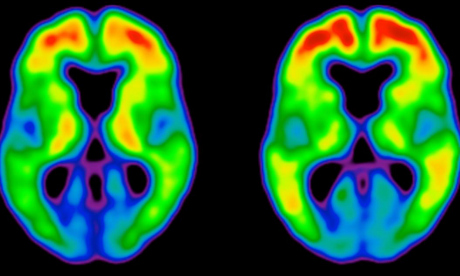
TAMMIE BENZINGER & TYLER BLAZEY, WASHINGTON UNIVERSITY
Patients with preclinical (centre) and clinical (right) Alzheimer's disease show a marked increase in amyloid plaques (red and yellow) in the brain.
Kang, who was a graduate student in neurologist David Holtzman's lab at Washington University in St Louis, Missouri, at the time, then made a further discovery: depriving the mice of sleep led to a dramatic rise in amyloid-β concentration1. “Once we saw that amyloid-β was going up and down with the sleep–wake cycle, the implications began to unfold,” says Holtzman. These findings suggested that sleep disturbances might actually precipitate plaque formation. And if a sleep deficit could increase the concentration of soluble amyloid-β, says Holtzman, then sleep abnormalities in earlier life may predispose people to Alzheimer's.
Holtzman's team went on to show that the natural cycle of waking and sleeping breaks down in mice following plaque formation, but is restored again when antibodies are used to eliminate the plaques2.
This phenomenon is not limited to mice. In people carrying mutations in the presenilin genes, which are strongly linked to the early onset, inherited form of Alzheimer's, Holtzman found that amyloid-β concentration in the cerebrospinal fluid fluctuates to a daily rhythm. He also showed that this cycle breaks down following plaque formation — and before any cognitive symptoms appear2.
So sleep disturbances might be one of the earliest manifestations of Alzheimer's disease, raising the tantalizing possibility of early intervention to help prevent or slow the inevitable march into the cognitive fog.
Early signs
Sleep disturbances might be an early sign of other neurodegenerative conditions as well. Last year, Roxanne Sterniczuk, a neuroscientist at Dalhousie University in Halifax, Canada, looked at data collected from approximately 14,600 people as part of the Survey of Health, Ageing and Retirement in Europe (SHARE), a long-term study of people aged 50 and over across 12 countries.
People who experienced sleep disturbances were more likely to be diagnosed with Alzheimer's within two to four years, according to the SHARE data. In particular, increased daytime sleepiness was the best predictor of the disease. The data also showed that daytime fatigue, a sleep disturbance that leads to exhaustion during the day, predicted those who went on to develop Parkinson's disease.
Parkinson's involves the progressive degeneration of midbrain neurons that produce dopamine — a neurotransmitter involved in regulating the sleep–wake switch. The drugs amphetamine and modafinil, for example, increase wakefulness by increasing dopamine signalling. Conversely, even one night of sleep deprivation reduces the number of dopamine receptors in the striatum, a region of the basal ganglia that is severely affected by the disease. Thus the sleep disturbances associated with Parkinson's may be caused by the degeneration of dopaminergic midbrain neurons.
Sterniczuk plans to analyse the SHARE data again to see if there are subtle differences between the sleep disturbances in presymptomatic Alzheimer's and Parkinson's patients. “If we can characterize the changes before symptoms appear then we can use them as diagnostic markers,” she says. “That might permit earlier treatment.”
The evidence is building for a correlation between disturbed sleep and neurodegenerative diseases, but the next step — discovering whether sleep disturbances are a cause of these conditions — will take considerably more research. In particular, establishing a causal relationship will require longitudinal studies that assess the sleeping patterns of large numbers of people over long periods of time, and link specific types of sleep disorder with the incidence of each disease. But to diagnose the diseases accurately, researchers must look for the tell-tale signs in the brains of study participants.
The Memory and Aging Project, run by David Bennett of Rush University Medical Center in Chicago, Illinois, will allow researchers to do that. The study involves 1,200 elderly people who will undergo annual neurological and psychiatric assessments and have agreed to donate their brains for research. Participants wear wrist monitors that record daily patterns of movement, revealing the times they sleep and wake, and their circadian rhythms. “We'll look at circuitry in the hypothalamus that's involved in sleep and circadian rhythms,” says Clifford Saper, a neurologist at Harvard Medical School in Boston, Massachusetts, who is collaborating on the project. “Presumably, neurodegenerative diseases disrupt sleep and circadian rhythms by damaging this circuitry.”
Testing this hypothesis in humans would be difficult, as it would involve comparing two groups of patients with sleep disorders who are at risk of a neurodegenerative disease, but treating only one group for their sleep problems. “It wouldn't be ethical to withhold treatment from patients, so we'd need to examine how circadian disruption affects neurodegeneration in an Alzheimer's mouse strain,” says Saper. “This would take a couple of years, but it may not mimic what happens in humans.”
Body clock
Sterniczuk is doing just that. In her Alzheimer's model, mice usually develop amyloid-β plaques at about 6 months of age and tau tangles — the other hallmark of Alzheimer's — at 12 months. Before then, their patterns of activity for a particular time of day, such as sleeping and feeding, differ from their non-pathogenic counterparts. The Alzheimer's mice also have fewer neurons in the suprachiasmatic nucleus (SCN), the part of the hypothalamus that regulates the circadian rhythm — a deficiency that makes their circadian clocks go off kilter3.
It's not yet clear whether these changes are associated with altered gene expression in the neurons of the SCN and the raphe nucleus, another part of the brain region that regulates sleep. “I'm trying to find out if changes in the animals' sleep rhythms are associated with altered protein levels in these regulatory regions,” Sterniczuk says. If they are, it raises the possibility of gene therapy. “Targeting these genes may alleviate the sleep and circadian-related symptoms, which may in turn slow the progression of cognitive decline.”
Fruitflies are also yielding clues to the link between neurodegeneration and circadian rhythms. In 2012, researchers at Oregon State University mutated the period gene, which plays a central role in governing the daily circadian rhythm, in fruitflies carrying a mutation in the sniffer gene, which causes neurodegeneration. The flies with both mutations degenerated more quickly, and had shorter lifespans, than flies with either mutation alone, suggesting that disrupted circadian rhythms can accelerate the process of neurodegeneration4.
The findings in mice and flies seem to be directly relevant to humans. In 2005, Jenny Morton, a neurophysiologist at the University of Cambridge, UK, reported that patients with Huntington's disease — a genetic disorder that affects muscle coordination and leads to cognitive decline and psychiatric problems — also have disrupted day–night activity patterns5. Moreover, this disturbance is accompanied by marked falls in the expression of two circadian clock genes in a mouse model of the disease.
Expression of the mouse clock gene mPer2 normally peaks in the morning and dips at night, whereas mBmal1 follows the opposite pattern, being expressed most at night and least in the morning. But in mice carrying genetic mutations associated with Huntington's disease, the expression profiles of both genes break down. In these mice, mPer2 expression is lowest in the afternoon, and the rhythmic expression of mBmal1 ceases altogether. What's more, the expression of both mPer2 and mBmal1 is disrupted in the parts of the brain that first succumb to Huntington's disease: the motor cortex and the striatum. These changes in gene expression are probably a result of disrupted circadian rhythms, rather than a cause of it. “We still don't know whether circadian changes are part of these diseases,” says Morton. “But if they do turn out to exacerbate the neurodegenerative process, then targeting sleep could be of potential therapeutic benefit.”
Genetic variation in clock genes seems to affect people's circadian rhythms, so it may also influence their susceptibility to neurodegeneration. In 2012, for example, Saper's team identified several variations that affect the activity of the human clock gene PER1 and are associated with being either an early riser or a night owl6. “This traces the tendency to be an early riser to a single gene, but we don't know what the implications are for neurodegeneration,” he says.
Broken dreams
Sleeping difficulties and neurodegeneration seem to reinforce one another in a vicious cycle. “Abnormal sleep in mid-life might cause protein aggregation that starts the disease off,” Holtzman says, “and the damage that causes may further disrupt sleep.”
“A lot of early data suggest that modifying sleep could actually delay the onset of disease.”
But could that dynamic be reversed? If disrupted sleep can predispose people to neurodegeneration, then might healthy sleeping patterns help protect the brain against it? Holtzman's group is testing this idea — essentially, whether it is possible to turn the vicious cycle virtuous. “A lot of early data suggest that modifying sleep could actually delay the onset of disease,” he says. “I think that's where the field should be going now, but it's not trivial translating all the animal studies directly into people.”
Holtzman is not alone in thinking this. Earlier this year, Alpar Lazar, who studies sleep and neurodegenerative disease at the University of Cambridge, presented preliminary data showing that bad sleep can precede the onset of Parkinson's disease by many years. His team found that sleep disturbances — particularly fragmented REM sleep — were more severe in patients who are nearer to disease onset than in those who are further away, who in turn had more disturbed sleep than healthy controls.
Huntington's disease is associated with mutations in the huntingtin gene, which contains a short repetitive DNA sequence called a CAG repeat. This repeat sequence is longer in Huntington's patients, leading to the production of misfolded Huntingtin protein. The longer the repeated sequence is, the earlier the disease begins, and the more severe its symptoms. Lazar and colleagues found that longer CAG repeats are also associated with worse sleep disturbances. “Our assumption is that sleep disturbances appear long before disease onset and precipitate disease progression,” says Lazar. So, he says, “intervening to improve sleep may slow down the disease process.” He is conducting a follow-up study to test this hypothesis.
There is already some evidence for this approach. Sleep apnoea, which is characterised by abnormally shallow and interrupted breathing, may almost double the risk of dementia7. Sleep apnoea is treatable, so early intervention could delay the onset not only of neurodegeneration, but also of normal age-related cognitive decline. “Once it is corrected, patients are much brighter and have better memory function,” says Saper, “but it's still not clear whether sleep loss itself increases neurodegeneration.”
Nevertheless, once clinicians wake up to the fact that sleep is so intimately linked to the most common neurodegenerative diseases, they may be in a better position to detect these debilitating neurological conditions at an early stage, and perhaps stop them in their tracks.



 Memory problems are a very commonly reported symptom of andropause. As with memory loss associated with menopause, it is important to be aware that occasional minor lapses in memory are nothing to worry about.
Memory problems are a very commonly reported symptom of andropause. As with memory loss associated with menopause, it is important to be aware that occasional minor lapses in memory are nothing to worry about.